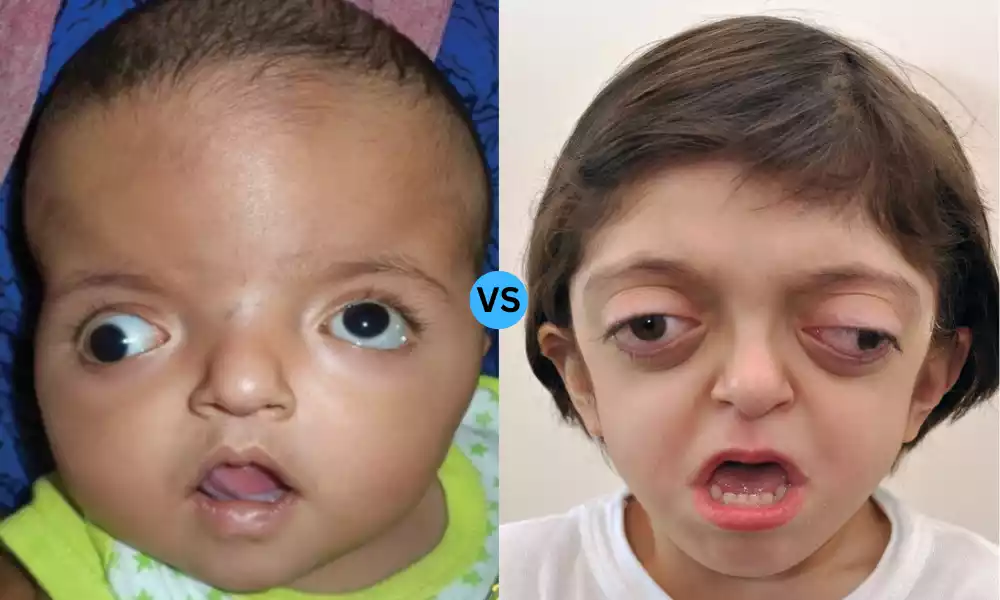
Apert and Crouzon Syndrome are genetic disorders that primarily affect the development of the skull and face, falling under the category of craniosynostosis syndromes. Apert syndrome, caused by mutations in the FGFR2 gene, is characterized by premature fusion of skull bones, facial abnormalities, and often fused fingers and toes (syndactyly). Crouzon syndrome, stemming from mutations in the FGFR2 or FGFR3 genes, also features skull and facial deformities but typically lacks the limb abnormalities seen in Apert syndrome. Both conditions usually require surgical intervention and a multidisciplinary approach for management.
What is Apert Syndrome?
Apert syndrome is a genetic disorder that primarily affects the development of the skull, face, and limbs. It is one of several craniosynostosis syndromes, which involve the premature fusion of the sutures between the bones of the skull.
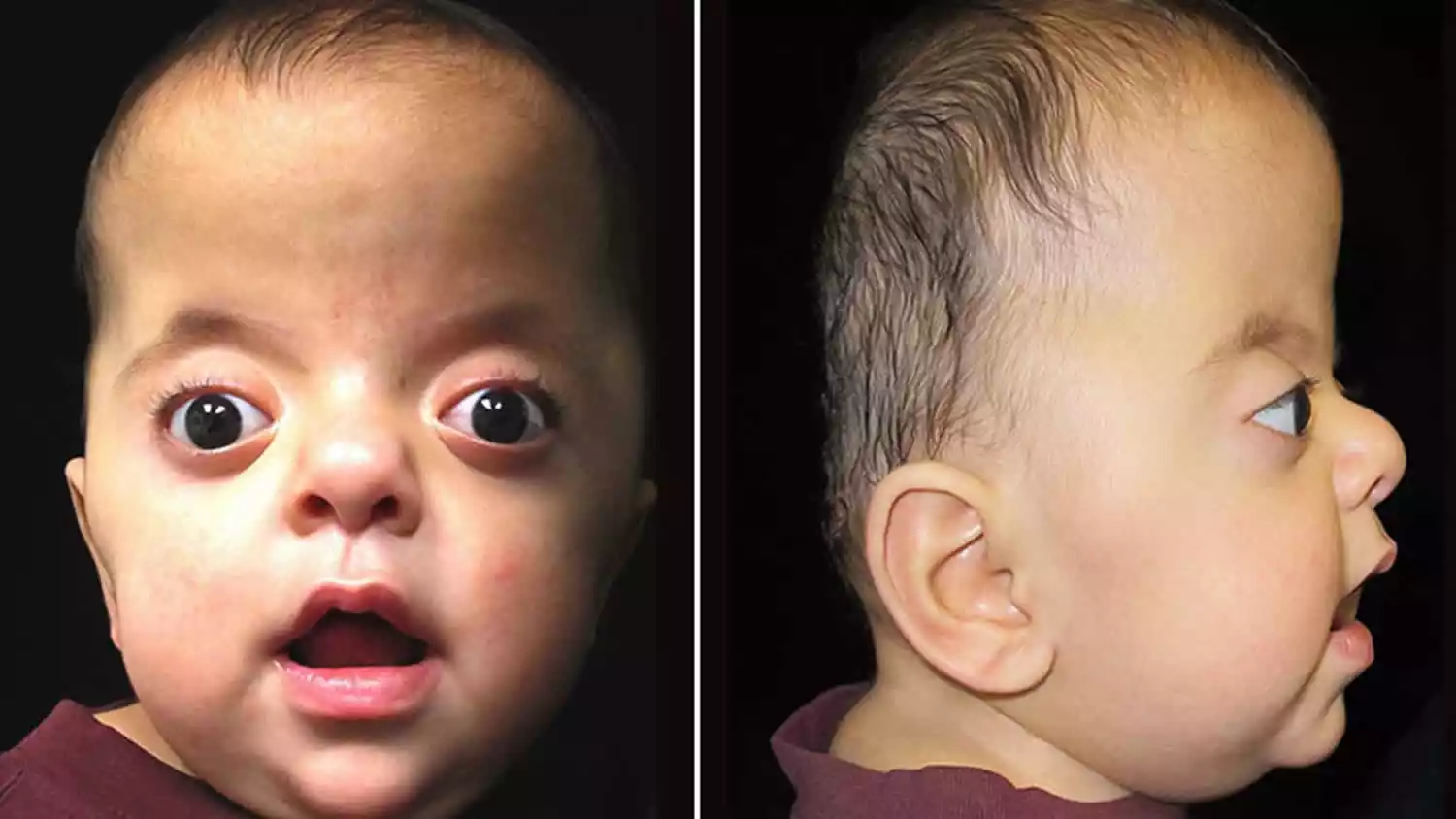
This early fusion prevents the skull from growing normally, leading to an abnormally shaped head and potential pressure on the developing brain. Apert syndrome is caused by mutations in the FGFR2 (Fibroblast Growth Factor Receptor 2) gene and is inherited in an autosomal dominant manner, although many cases result from spontaneous mutations.
Key features of Apert syndrome include:
- Craniosynostosis: Premature fusion of skull bones, resulting in an abnormal head shape.
- Syndactyly: Fusion of fingers and/or toes, often requiring surgical separation.
- Facial Abnormalities: These may include shallow eye sockets, a beaked nose, and an underdeveloped upper jaw.
- Cognitive and Developmental Delays: While intellectual capabilities can vary widely, some individuals experience developmental delays.
Diagnosis is generally made based on physical characteristics at birth or through prenatal imaging like ultrasounds. Genetic testing can confirm the diagnosis. Treatment often involves surgical intervention to correct the skull deformities and separate fused digits, as well as other supportive therapies like speech and occupational therapy.
It’s essential to consult healthcare professionals for diagnosis and treatment, as the condition requires a multidisciplinary approach for optimal management.
What is Crouzon Syndrome?
Crouzon syndrome is a genetic disorder characterized by the premature fusion of certain skull bones, known as craniosynostosis. This early fusion disrupts the normal growth pattern of the skull and can affect the shape of the head and face.
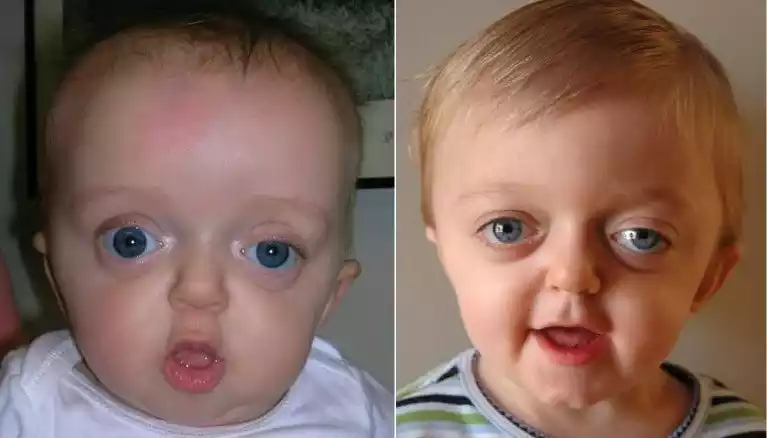
The syndrome is generally caused by mutations in the FGFR2 (Fibroblast Growth Factor Receptor 2) or FGFR3 (Fibroblast Growth Factor Receptor 3) genes and is typically inherited in an autosomal dominant manner, though sporadic mutations can also occur.
Key features of Crouzon syndrome include:
- Craniosynostosis: The premature fusion of the skull bones results in an abnormal head shape and may exert pressure on the brain.
- Facial Abnormalities: These often include shallow eye sockets, which can make the eyes appear to bulge; a beaked nose; and an underdeveloped upper jaw.
- Eye Problems: Due to the shallow eye sockets, issues like strabismus (misaligned eyes) and other vision problems are common.
- No Limb Involvement: Unlike some other craniosynostosis syndromes like Apert syndrome, Crouzon syndrome usually does not involve fusion of the fingers or toes (syndactyly).
Diagnosis is generally made through physical examination at birth or shortly thereafter, although prenatal imaging can sometimes detect features of the syndrome. Genetic testing can be used to confirm the diagnosis.
Treatment often involves surgical procedures to correct the cranial and facial deformities, with the aim to alleviate pressure on the brain and improve cosmetic appearance. Ophthalmological and orthodontic interventions may also be required.
As with other craniosynostosis syndromes, Crouzon syndrome typically requires a multidisciplinary approach to treatment, involving specialists like pediatricians, neurosurgeons, ophthalmologists, and orthodontists.
Commonalities Between Apert and Crouzon Syndrome
Apert and Crouzon syndromes share several common features, primarily because they both fall under the category of craniosynostosis syndromes. Here are some commonalities between the two:
- Genetic Origin: Both syndromes are typically caused by mutations in the FGFR2 (Fibroblast Growth Factor Receptor 2) gene, although Crouzon can also be caused by mutations in the FGFR3 gene.
- Craniosynostosis: Both conditions feature the premature fusion of skull bones, which can result in abnormal head shapes and potentially exert pressure on the brain.
- Facial Abnormalities: Both syndromes can lead to shallow eye sockets, a beaked nose, and underdeveloped jaws, among other facial deformities.
- Autosomal Dominant Inheritance: Both conditions are usually inherited in an autosomal dominant manner, meaning that only one copy of the altered gene is sufficient to cause the disorder. However, many cases are also due to spontaneous mutations.
- Surgical Treatment: Treatment for both conditions often involves surgical intervention to correct skull and facial abnormalities, usually performed in early childhood to minimize complications.
- Multi-disciplinary Care: Both syndromes require a comprehensive, multidisciplinary approach to treatment, involving a team of specialists such as pediatricians, neurosurgeons, ophthalmologists, and orthodontists, among others.
- Diagnosis: Both conditions are often diagnosed shortly after birth based on physical characteristics, although prenatal imaging like ultrasounds can sometimes detect them. Genetic testing can be used to confirm the diagnosis.
- Potential for Developmental Delays: While the extent can vary among individuals, both conditions may be associated with developmental and cognitive delays, partly due to the impact of craniosynostosis on brain development.
- Ophthalmological Issues: Due to the facial and cranial deformities, both syndromes can lead to eye problems like strabismus or vision issues that may require intervention.
Despite these commonalities, it’s essential to note that each syndrome has its own distinct features and complications. For instance, Apert syndrome often includes limb abnormalities like syndactyly, which is usually absent in Crouzon syndrome. Always consult healthcare professionals for diagnosis and treatment.
Comparison Table of Apert and Crouzon Syndrome
Certainly, a comparison table can help clarify the similarities and differences between Apert and Crouzon syndromes:
| Feature | Apert Syndrome | Crouzon Syndrome |
|---|---|---|
| Genetic Origin | FGFR2 gene mutations | FGFR2 or FGFR3 gene mutations |
| Inheritance | Autosomal Dominant | Autosomal Dominant |
| Craniosynostosis | Yes | Yes |
| Facial Abnormalities | Shallow eye sockets, beaked nose, underdeveloped jaw | Shallow eye sockets, beaked nose, underdeveloped jaw |
| Syndactyly (Fused Fingers/Toes) | Yes | No |
| Eye Problems | Common (e.g., strabismus) | Common (e.g., strabismus) |
| Developmental Delays | Possible | Possible |
| Diagnosis Method | Physical examination, genetic testing | Physical examination, genetic testing |
| Treatment | Surgery for skull and facial abnormalities, syndactyly correction, supportive therapies | Surgery for skull and facial abnormalities, supportive therapies |
| Specialist Involvement | Pediatricians, neurosurgeons, orthodontists, ophthalmologists, etc. | Pediatricians, neurosurgeons, orthodontists, ophthalmologists, etc. |
This table is intended for educational purposes and should not replace professional medical advice. Always consult healthcare professionals for diagnosis and treatment.
Causes of Apert and Crouzon Syndrome
Both Apert syndrome and Crouzon syndrome are primarily caused by genetic mutations affecting the fibroblast growth factor receptors, specifically in the FGFR2 and/or FGFR3 genes. These receptors play a vital role in the development of bone and tissue.
Causes of Apert Syndrome
- Genetic Mutation: The majority of cases of Apert syndrome are caused by mutations in the FGFR2 gene.
- Autosomal Dominant Inheritance: In families where Apert syndrome is present, it is typically inherited in an autosomal dominant manner. This means that only one parent needs to carry the mutated gene for the child to inherit the disorder.
- Sporadic Cases: In many instances, Apert syndrome arises spontaneously due to a new mutation, with no previous family history of the disorder.
Causes of Crouzon Syndrome
- Genetic Mutation: Crouzon syndrome is primarily caused by mutations in the FGFR2 gene, although some cases can also be attributed to mutations in the FGFR3 gene.
- Autosomal Dominant Inheritance: Like Apert syndrome, Crouzon syndrome is usually inherited in an autosomal dominant pattern.
- Sporadic Cases: As with Apert syndrome, Crouzon syndrome can also occur spontaneously due to a new genetic mutation, without any family history.
The presence of these genetic mutations leads to the premature fusion of bones in the skull (craniosynostosis), affecting the normal development of the skull and often the face.
While genetic factors are the primary cause, the expression and severity of the syndromes can vary among individuals, even among those with the same genetic mutation.
Diagnosis Procedures of Apert and Crouzon Syndrome
Diagnosis procedures for Apert and Crouzon syndromes are quite similar due to the overlapping characteristics of these craniosynostosis syndromes. Here are some of the common diagnostic procedures used:
Physical Examination
Apert and Crouzon: A detailed physical examination shortly after birth often reveals the characteristic features of either syndrome, such as an abnormal skull shape and facial features.
Imaging Studies
- Ultrasound: In some cases, features of either syndrome may be detected prenatally through ultrasound imaging.
- X-rays and CT scans: These can provide a more detailed view of the skull, highlighting the extent of craniosynostosis and any facial abnormalities.
Genetic Testing
- Apert Syndrome: Genetic testing can identify mutations in the FGFR2 gene.
- Crouzon Syndrome: Testing can identify mutations in the FGFR2 or FGFR3 genes.
Other Diagnostic Tools
- Ophthalmic Examination: Due to the high likelihood of eye issues, a detailed eye examination may be part of the diagnostic process for both syndromes.
- Developmental Assessments: Cognitive and motor skills may be assessed, especially in children, to determine if there are any developmental delays.
Prenatal Diagnosis
- Amniocentesis or Chorionic Villus Sampling (CVS): For families with a history of either syndrome or who are known carriers of the relevant mutations, prenatal genetic testing may be an option.
Consultation with Specialists
For both syndromes, diagnosis is usually confirmed by a multi-disciplinary team of healthcare professionals including:
- Pediatricians
- Geneticists
- Neurosurgeons
- Ophthalmologists
- Orthodontists
- Radiologists
Due to the complexities and potential complications of these syndromes, a comprehensive approach involving multiple healthcare specialists is usually required for accurate diagnosis and effective management. It’s important to note that the above information is intended for educational purposes and should not replace professional medical advice. Always consult healthcare professionals for diagnosis and treatment.
Treatment for Apert and Crouzon Syndrome
The treatment for both Apert and Crouzon syndromes typically involves a multidisciplinary approach due to the complexity of the conditions. The main goal is to alleviate any pressure on the brain, improve facial appearance and function, and address any other associated complications. Here’s a breakdown of treatments for both syndromes:
Surgical Interventions:
- Cranial Vault Remodeling: This surgery reshapes the skull to alleviate any pressure on the brain and correct the abnormal head shape. It’s typically performed within the first year of life.
- Midface Advancement: As children with these syndromes often have an underdeveloped midface, a surgical procedure may be carried out to move the midface forward, improving breathing, bite alignment, and appearance.
- Orbital Surgery: Due to the shallow eye sockets, surgery might be necessary to correct the position and shape of the orbits, reducing the appearance of bulging eyes.
- Syndactyly Correction (specific to Apert syndrome): Surgery is done to separate fused fingers or toes, typically carried out in stages during the early years of life.
- Orthodontic and Dental Interventions: Malocclusion and dental crowding are common, so braces, extractions, or other orthodontic treatments might be necessary.
Other Treatments:
- Hearing Aids: Some individuals with these syndromes might have hearing issues, and hearing aids can be beneficial in these cases.
- Speech Therapy: Due to facial abnormalities, some children might develop speech difficulties and can benefit from speech therapy.
- Physical and Occupational Therapy: Especially in Apert syndrome, where there might be hand and foot abnormalities, therapy can help improve function and dexterity.
- Eye Care: Regular eye exams are crucial to monitor and manage any ophthalmological issues, including strabismus or vision problems.
- Counseling and Support: Living with a noticeable physical difference can be emotionally challenging. Counseling or support groups can offer emotional and psychological support to patients and their families.
Long-Term Monitoring:
Regular follow-up with specialists (like pediatricians, neurosurgeons, ophthalmologists, orthodontists, and others) is vital to monitor the child’s development, detect any complications early, and make necessary adjustments in treatment plans.
While surgical intervention is often essential, the timing, type, and sequence of surgeries can vary based on the severity of the condition, the child’s age, and other individual factors. It’s crucial to consult with a team of specialists to determine the best treatment plan tailored to each patient.
Latest Research and Developments
As of my last update in January 2022, research into craniosynostosis syndromes like Apert and Crouzon has been ongoing. Here are some areas of interest, though specific advancements after this date would require you to check current scientific journals or medical databases for the most recent information:
- Genetic Insights: Improved understanding of the FGFR2 and FGFR3 genes (as well as other potentially relevant genes) and the exact mechanisms by which mutations lead to the phenotypic expressions seen in Apert and Crouzon syndromes.
- Improved Surgical Techniques: As surgical interventions are primary treatments for these syndromes, there’s consistent effort in refining techniques to improve outcomes, reduce complications, and minimize the need for multiple procedures. This includes the use of endoscopic surgery and the development of custom-made implants for cranial vault remodeling.
- Stem Cell Research: Some researchers are exploring the potential of stem cells in treating craniosynostosis. This could involve using stem cells to regenerate affected skull bones without the need for invasive surgery.
- 3D Printing: The rise of 3D printing technology has potential applications in craniofacial surgery, allowing surgeons to create accurate models of the patient’s skull or even custom implants.
- Early Detection: Efforts are being made to refine prenatal screening techniques, allowing for earlier diagnosis and potentially even in-utero interventions in the future.
- Non-Surgical Treatments: Exploring medications or other non-surgical methods to prevent or treat the premature fusion of cranial sutures is a potential avenue of research.
- Patient Support and Rehabilitation: Research into effective rehabilitation strategies and support tools, especially for children with these syndromes, can enhance quality of life and long-term outcomes.
- Long-term Outcomes: Studies are being conducted to understand the long-term outcomes, both physical and psychological, of individuals with Apert and Crouzon syndromes to inform treatment strategies and patient support.
For the very latest in research and developments, checking databases like PubMed, resources from specialized institutions, and relevant conferences or seminars would provide up-to-date insights. If you are specifically concerned about these conditions, discussing with a genetic counselor or specialist would also be advisable.
Case Studies
Case studies provide valuable insights into the manifestations, treatment, and outcomes of particular medical conditions in real-world settings. However, to access specific, up-to-date case studies on Apert or Crouzon syndromes, you would typically look into scientific journals, medical databases, or conference proceedings.
As of my last update in January 2022, I don’t have the capability to provide real-time or very recent case studies. I can provide a general outline of how such a case study might be structured based on the information available:
Case Study: Early Intervention in a Child with Apert Syndrome
Introduction: Brief description of Apert syndrome, emphasizing its rarity and genetic basis.
Patient Presentation:
- Gender: Male
- Age at diagnosis: 6 months
- Initial Concerns: Parents noted an irregular shape to the baby’s skull and fused fingers and toes.
- Physical Examination: Abnormal head shape, fused sutures evident on palpation, syndactyly of both hands and feet.
Diagnostic Process:
- Genetic Testing: Confirmed mutation in the FGFR2 gene.
- CT scan: Showed premature fusion of coronal sutures.
Treatment:
- Surgical: At 8 months, the child underwent cranial vault remodeling to address craniosynostosis. At 1 year, the first surgery for syndactyly separation was conducted on the hands.
- Rehabilitative: Physical therapy introduced post-surgery to aid in hand function after syndactyly correction.
Follow-up & Outcome:
- At age 3: The child’s head shape was within normal range, with no signs of increased intracranial pressure. Further surgeries for syndactyly correction in the feet are planned. Developmentally, the child is showing some delays in fine motor skills, but with therapy, significant improvements are noted.
- Continuous monitoring is planned with a multi-disciplinary team.
Early diagnosis and intervention in Apert syndrome can lead to improved physical outcomes and developmental trajectory. Multi-disciplinary care is essential for comprehensive management.
Conclusion
Apert and Crouzon syndromes are both rare genetic disorders characterized by the premature fusion of cranial sutures, known as craniosynostosis, leading to abnormalities in the skull and face. While both conditions arise from mutations in the FGFR genes, Apert syndrome is also marked by syndactyly, or the fusion of fingers and toes.
Diagnosis primarily involves physical examination, imaging studies, and genetic testing. Treatment is multidisciplinary, with surgical interventions playing a central role in managing the craniofacial abnormalities. Both conditions highlight the importance of early detection and comprehensive care for optimal outcomes.