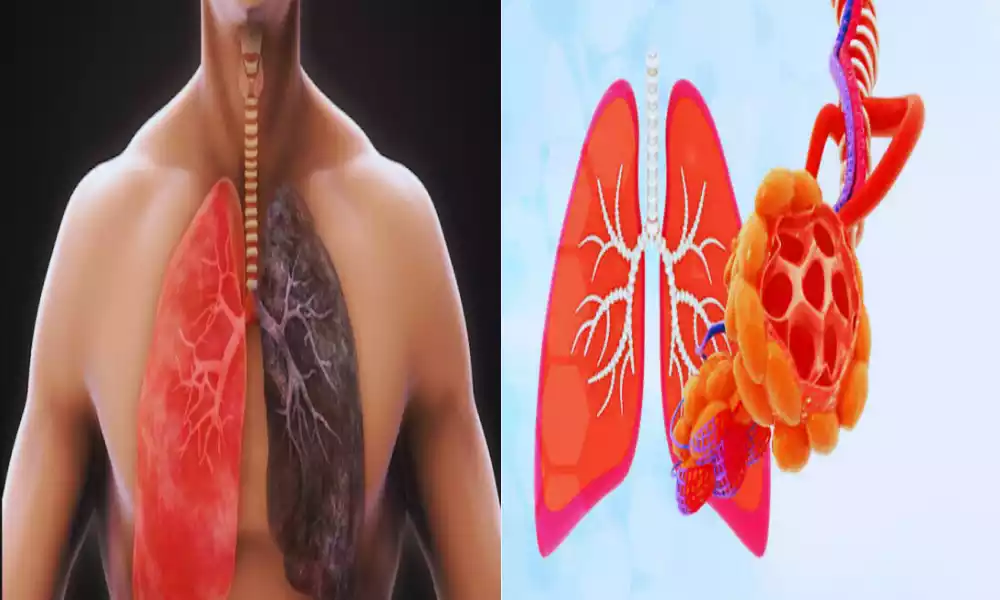
Interstitial Lung Disease and Pulmonary Fibrosis are respiratory conditions that affect the lungs, often leading to breathing difficulties and reduced lung function. ILD refers to a group of disorders that impact the interstitial tissue surrounding the air sacs, while PF is a specific type of ILD characterized by lung scarring. These conditions can be caused by various factors, and early diagnosis and effective management are crucial for individuals dealing with ILD and PF. In this article, we will delve deeper into their causes, symptoms, diagnosis, treatment options, and ways to improve the quality of life for those affected.
Definition of Interstitial Lung Disease
Interstitial Lung Disease (ILD), also known as interstitial pulmonary fibrosis, is a broad category of lung disorders characterized by inflammation and scarring (fibrosis) of the interstitium, which is the tissue that surrounds the air sacs (alveoli) in the lungs. The interstitium plays a crucial role in supporting the structure of the lungs and facilitating the exchange of oxygen and carbon dioxide during respiration.
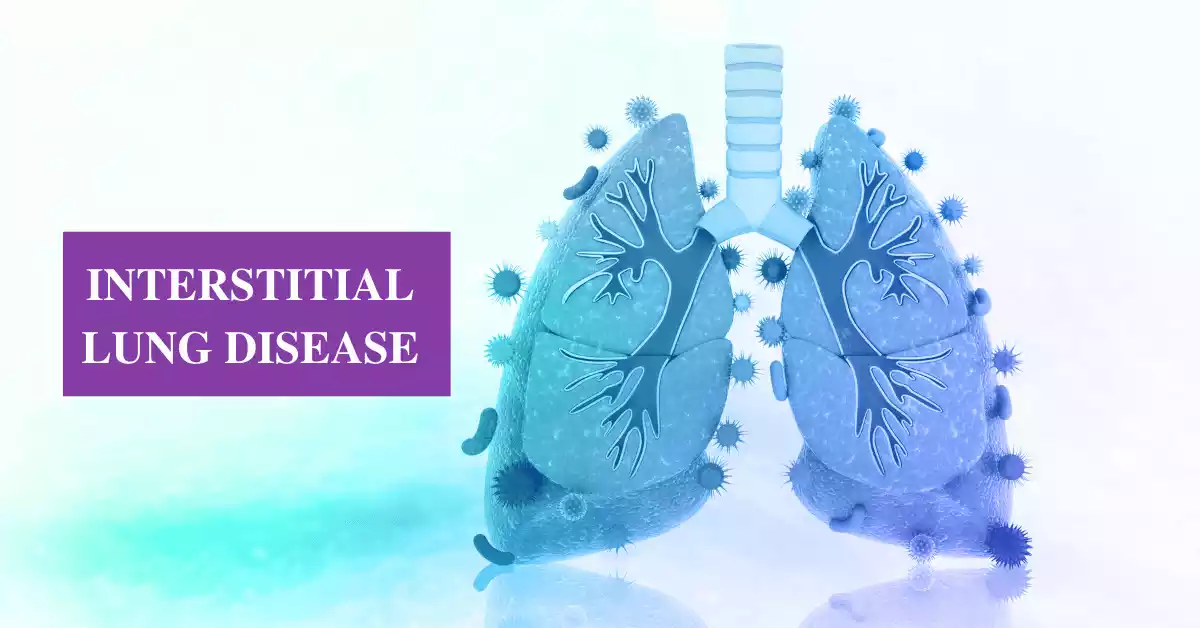
ILD encompasses a wide range of specific lung conditions that affect the interstitial tissue, leading to impaired lung function. These conditions may have various causes, including environmental exposures, autoimmune diseases, infections, and genetic factors. Common symptoms of ILD include shortness of breath, cough, and reduced exercise tolerance. Accurate diagnosis and proper management are essential to mitigate the progression of these diseases and improve patients’ quality of life.
Symptoms of Interstitial Lung Disease
Interstitial Lung Disease (ILD) encompasses a diverse group of lung disorders, and the symptoms can vary depending on the specific subtype and the extent of lung involvement. However, there are some common symptoms that individuals with ILD may experience. These symptoms typically develop gradually and can worsen over time. Common symptoms of ILD include:
- Shortness of Breath (Dyspnea): This is one of the most prevalent symptoms of ILD. Initially, it may occur with physical activity but can progress to the point where it occurs even at rest.
- Persistent Dry Cough: Many individuals with ILD develop a chronic, non-productive cough that doesn’t produce mucus or phlegm.
- Fatigue: Fatigue is a common complaint among ILD patients and can result from the increased effort required to breathe.
- Reduced Exercise Tolerance: Due to the limited lung function, individuals with ILD often experience a decreased ability to engage in physical activities or exercise.
- Chest Discomfort: Some people with ILD report chest discomfort or a sense of tightness in the chest.
- Clubbing of Fingers and Toes: In some ILD cases, the fingers and toes may develop clubbing, a condition where the fingertips and nail beds become enlarged and rounded.
- Unintentional Weight Loss: Progressive ILD can lead to weight loss, primarily due to the increased energy expenditure associated with labored breathing.
- Wheezing: Wheezing or a high-pitched whistling sound when breathing may occur in some ILD cases.
- Cyanosis: In advanced stages, individuals may develop cyanosis, a bluish tint to the skin and lips, indicating low oxygen levels in the blood.
The severity and combination of symptoms can vary widely among ILD patients, and not everyone will experience all of these symptoms. Additionally, ILD symptoms can overlap with those of other respiratory conditions, making accurate diagnosis and differentiation crucial.
If you or someone you know is experiencing persistent respiratory symptoms, especially if they worsen over time, it’s important to seek medical evaluation. Early diagnosis and appropriate management can help improve the quality of life for individuals with ILD.
Definition of Pulmonary Fibrosis
Pulmonary fibrosis (PF) is a specific type of interstitial lung disease characterized by the progressive scarring (fibrosis) of the lung tissue, particularly the interstitium. This fibrosis results in the thickening and stiffening of the lung’s interstitial tissue, which can lead to reduced lung function and impaired oxygen exchange. Over time, as the scarring worsens, it becomes increasingly difficult for the affected individual to breathe properly.
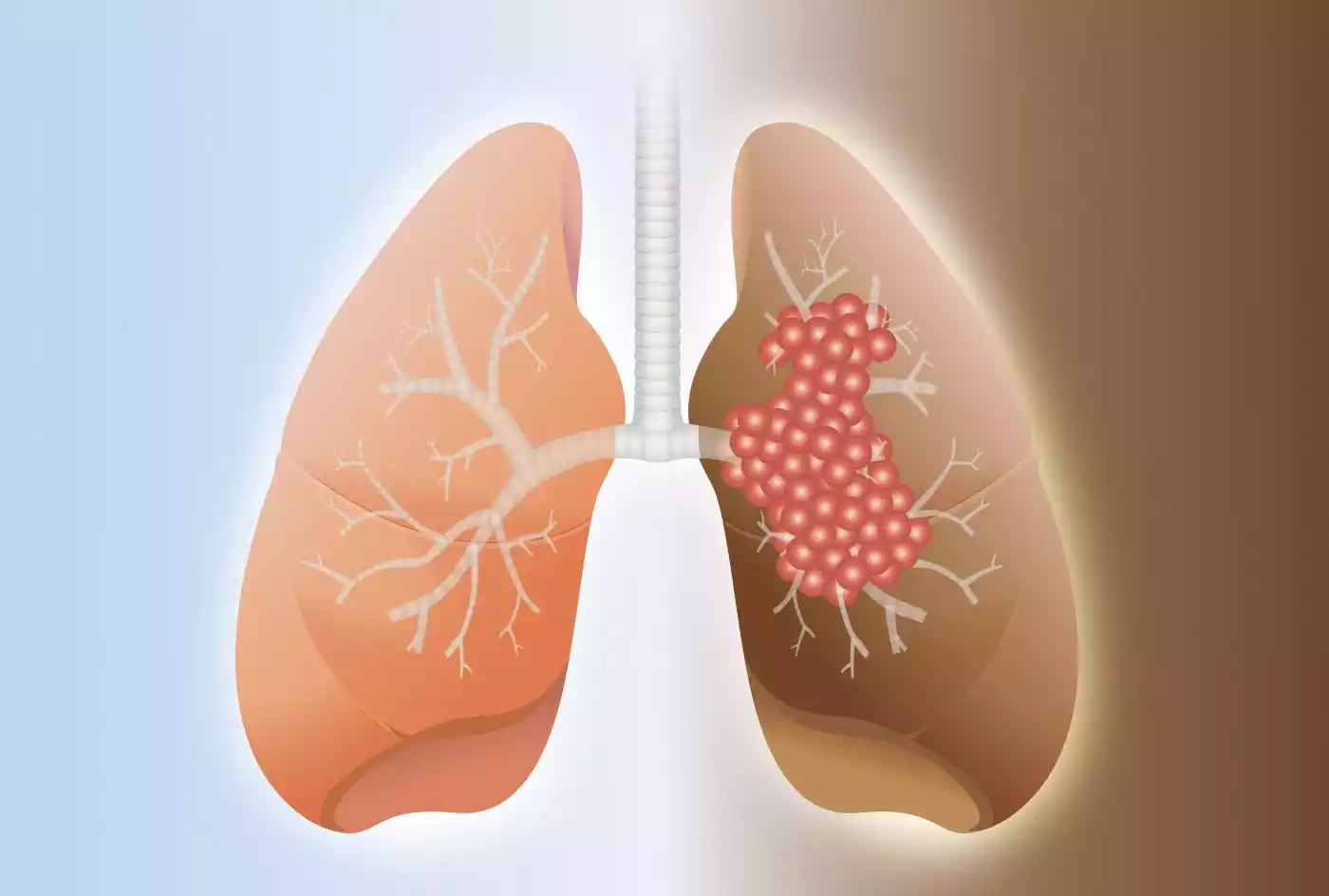
Pulmonary fibrosis can have various underlying causes, including environmental factors, occupational exposures, certain medications, connective tissue diseases, and in some cases, it may be of unknown origin, referred to as idiopathic pulmonary fibrosis (IPF). Symptoms of pulmonary fibrosis typically include shortness of breath, a persistent dry cough, fatigue, and reduced exercise tolerance.
While there is currently no cure for PF, various treatment options, including medications and lung transplantation, may help manage symptoms and slow the progression of the disease. Early diagnosis and intervention are crucial for optimizing the quality of life for individuals with pulmonary fibrosis.
Symptoms of Pulmonary Fibrosis
Pulmonary fibrosis (PF) is a progressive lung disease characterized by the scarring (fibrosis) of lung tissue, particularly the interstitium. The symptoms of PF can vary in severity from person to person, and they often develop gradually over time. Common symptoms of pulmonary fibrosis include:
- Shortness of Breath (Dyspnea): This is one of the hallmark symptoms of PF. It usually starts with exertional dyspnea (shortness of breath during physical activity) but can progress to breathlessness even at rest as the disease advances.
- Persistent Dry Cough: Many individuals with PF develop a chronic, dry cough that does not produce mucus or phlegm. This cough can be irritating and persistent.
- Fatigue: PF can lead to fatigue, which may be both physical and mental. The effort required to breathe can contribute to a sense of tiredness.
- Reduced Exercise Tolerance: People with PF often notice a decreased ability to engage in physical activities or exercise due to shortness of breath.
- Chest Discomfort: Some individuals with PF report chest discomfort or a feeling of tightness in the chest, particularly during periods of increased breathlessness.
- Unintentional Weight Loss: Progressive PF can lead to weight loss, primarily due to the increased energy expenditure associated with labored breathing.
- Clubbing of Fingers and Toes: In some cases, PF can cause clubbing, a condition in which the fingertips and nail beds become enlarged and rounded.
- Wheezing: Wheezing or a high-pitched whistling sound when breathing may occur in some PF cases.
- Cyanosis: In advanced stages of PF, individuals may develop cyanosis, a bluish tint to the skin and lips, indicating low oxygen levels in the blood.
- Difficulty in Taking Deep Breaths: Patients with PF often find it challenging to take deep breaths or fully expand their lungs, which can contribute to feelings of breathlessness.
- Respiratory Infections: PF can increase the risk of respiratory infections, such as pneumonia, due to compromised lung function and impaired ability to clear mucus from the airways.
While these are common symptoms of PF, not all individuals will experience all of them, and the severity and combination of symptoms can vary. Early diagnosis and appropriate management are essential to slow the progression of the disease and improve the quality of life for individuals with pulmonary fibrosis. If you or someone you know is experiencing persistent respiratory symptoms, especially if they worsen over time, it’s important to seek medical evaluation and consultation with a healthcare professional.
Comparison Table of Interstitial Lung Disease and Pulmonary Fibrosis
Here’s a comparison table of Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF) to highlight their key differences and similarities:
| Aspect | Interstitial Lung Disease (ILD) | Pulmonary Fibrosis (PF) |
|---|---|---|
| Definition | A broad category of lung disorders characterized by inflammation and fibrosis of the lung’s interstitial tissue. | A specific type of ILD, characterized by progressive scarring (fibrosis) of the lung tissue, particularly the interstitium. |
| Etiology and Causes | Diverse causes, including environmental exposures, autoimmune diseases, infections, and genetic factors. | Various underlying causes, including environmental factors, occupational exposures, medications, connective tissue diseases, and idiopathic (unknown origin). |
| Pathophysiology | Inflammation and fibrosis primarily affect the interstitium, the lung’s supporting tissue. | Progressive scarring of the lung tissue leads to thickening and stiffening of the interstitial tissue. |
| Clinical Presentation and Symptoms | Common symptoms include shortness of breath, cough, reduced exercise tolerance, and chest discomfort. | Typical symptoms include shortness of breath, persistent dry cough, fatigue, and decreased exercise capacity. |
| Diagnostic Approaches | Diagnosis involves imaging (HRCT, chest X-ray), pulmonary function tests, and sometimes lung biopsy. | Similar diagnostic methods as ILD, including imaging, pulmonary function tests, and biopsy. |
| Treatment and Management Strategies | Treatment options include medications to reduce inflammation and manage symptoms, oxygen therapy, and pulmonary rehabilitation. | Treatment may include medications (e.g., anti-fibrotic drugs), lung transplantation for severe cases, and palliative care. |
| Prognosis and Outlook | Prognosis varies depending on the specific ILD subtype and its severity. | Prognosis depends on the underlying cause and extent of fibrosis; it can be challenging, particularly in advanced stages. |
| Common Subtypes | Various subtypes, including idiopathic pulmonary fibrosis (IPF), hypersensitivity pneumonitis, sarcoidosis, and more. | Pulmonary fibrosis is itself a subtype of ILD. Common subtypes include IPF, connective tissue disease-associated PF, and others. |
| Importance of Early Diagnosis | Early diagnosis is crucial for timely intervention and better disease management. | Early diagnosis is essential to slow the progression of fibrosis and improve the quality of life for patients. |
This table provides an overview of the key distinctions and similarities between Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF), emphasizing their definitions, causes, clinical features, diagnostic methods, treatments, and prognosis.
Similarities between Interstitial Lung Disease and Pulmonary Fibrosis
Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF) share several similarities due to the fact that PF is a specific type of ILD. Here are some of the commonalities between these two conditions:
- Affect Interstitial Tissue: Both ILD and PF primarily involve the interstitial tissue of the lungs. They result in scarring (fibrosis) and inflammation within this supportive lung structure.
- Respiratory Symptoms: Patients with both ILD and PF typically experience similar respiratory symptoms, such as shortness of breath, a persistent dry cough, and reduced exercise tolerance.
- Diagnostic Approaches: The diagnostic approaches for ILD and PF overlap significantly. Physicians use imaging techniques like high-resolution computed tomography (HRCT) and chest X-rays, pulmonary function tests, and sometimes lung biopsies to diagnose and differentiate these conditions.
- Treatment Strategies: Although the specific treatment approaches may vary depending on the underlying cause and subtype, both ILD and PF can be managed with similar strategies. These include medications to reduce inflammation and slow fibrosis progression, oxygen therapy to improve oxygen levels, and pulmonary rehabilitation to enhance lung function and overall well-being.
- Prognosis Variability: Both ILD and PF can have variable prognoses depending on factors like the subtype, the extent of lung involvement, and the patient’s overall health. Some forms of ILD and PF can progress slowly, while others may progress more rapidly.
- Importance of Early Diagnosis: Early diagnosis and intervention are crucial for both ILD and PF. Timely recognition and treatment can help slow disease progression and improve the patient’s quality of life.
- Supportive Care: Patients with ILD and PF often require similar forms of supportive care, including supplemental oxygen, physical therapy, and palliative care to manage symptoms and enhance comfort.
- Chronic Nature: Both ILD and PF are typically chronic conditions that require ongoing management and monitoring by healthcare professionals.
PF is a subset of ILD, and the term “pulmonary fibrosis” is often used to describe a specific type of interstitial lung disease characterized by progressive fibrosis. Therefore, while they share many similarities, PF can be considered a more specific diagnosis within the broader category of ILD.
Diagnosis of Interstitial Lung Disease and Pulmonary Fibrosis
Diagnosing Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF) typically involves a combination of clinical evaluation, imaging studies, pulmonary function tests, and, in some cases, lung biopsy. Here’s an overview of the diagnostic process for ILD and PF:
- Medical History and Physical Examination:
- The healthcare provider will take a detailed medical history, including any symptoms, risk factors, occupational exposures, and relevant medical conditions.
- A physical examination may reveal signs such as clubbing of fingers, abnormal breath sounds, and cyanosis.
- Imaging Studies:
- High-Resolution Computed Tomography (HRCT): This imaging technique provides detailed pictures of the lungs and is often the first-line tool for diagnosing ILD and PF. HRCT can reveal characteristic patterns of lung abnormalities.
- Chest X-ray: While less sensitive than HRCT, a chest X-ray can provide an initial assessment of lung changes.
- Pulmonary Function Tests (PFTs):
- PFTs assess lung function and may include spirometry and diffusion capacity tests. These tests can help determine the severity of lung impairment and the extent of damage.
- Blood Tests:
- Blood tests may be conducted to check for specific autoimmune markers, assess oxygen levels, and rule out other conditions that can mimic ILD or PF.
- Lung Biopsy:
- In cases where the diagnosis remains uncertain after initial evaluation, a lung biopsy may be necessary. This involves taking a small tissue sample from the lung for microscopic examination. There are different biopsy techniques, including surgical lung biopsy and bronchoscopy with transbronchial biopsy.
- Bronchoalveolar Lavage (BAL):
- BAL is a procedure where a small amount of saline solution is instilled into a lung segment and then retrieved for analysis. It can help identify infections or other causes of lung inflammation.
- Multidisciplinary Team Evaluation:
- In many ILD and PF cases, a multidisciplinary team, including pulmonologists, radiologists, pathologists, and rheumatologists, may collaborate to review clinical and diagnostic data, ensuring a comprehensive assessment.
- Specialized Testing:
- Depending on the suspected cause, additional tests such as autoimmune serologies, genetic testing, or environmental exposure assessments may be necessary.
- Monitoring and Follow-Up:
- ILD and PF are often chronic and progressive conditions, so regular follow-up visits are crucial to monitor disease progression and adjust treatment strategies as needed.
Diagnosing ILD and PF can be challenging due to their heterogeneity and similarities to other respiratory conditions. A comprehensive evaluation, including clinical, radiological, and pathological assessments, is essential to arrive at an accurate diagnosis and develop a tailored treatment plan.
Treatment for Interstitial Lung Disease and Pulmonary Fibrosis
Treatment for Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF) aims to manage symptoms, slow disease progression, improve lung function, and enhance the patient’s quality of life. The specific treatment approach may vary depending on the underlying cause, the severity of the condition, and individual patient factors.
Here are common treatment strategies:
- Medications:
- Corticosteroids: In some ILD cases, corticosteroids like prednisone may be prescribed to reduce lung inflammation.
- Immunosuppressive Drugs: Medications like azathioprine, mycophenolate, or cyclophosphamide may be used in certain ILD types, especially when an autoimmune component is involved.
- Antifibrotic Drugs: For idiopathic pulmonary fibrosis (IPF), two antifibrotic drugs, nintedanib and pirfenidone, have been approved to slow disease progression.
- Oxygen Therapy:
- Supplemental oxygen is often prescribed to maintain adequate oxygen levels in the blood and alleviate breathlessness.
- Pulmonary Rehabilitation:
- Pulmonary rehabilitation programs include exercise training, education, and support to improve lung function, stamina, and overall well-being.
- Lung Transplantation:
- In advanced cases of ILD or PF with severe lung damage, lung transplantation may be considered as a treatment option.
- Symptom Management:
- Medications may be prescribed to manage specific symptoms, such as cough or acid reflux.
- Pulmonary hygiene techniques, such as postural drainage and percussion, may help clear mucus from the airways.
- Managing comorbidities, such as gastroesophageal reflux disease (GERD) or sleep apnea, is important to improve overall health.
- Vaccinations:
- Immunizations, including annual influenza vaccines and pneumococcal vaccines, are recommended to prevent respiratory infections that can exacerbate ILD or PF.
- Supportive Care:
- Supportive care focuses on improving the patient’s comfort and quality of life. This may include palliative care, nutritional support, and counseling.
- Lifestyle Changes:
- Smoking cessation is crucial for all ILD and PF patients to slow disease progression.
- Avoiding environmental exposures and hazards that may worsen the condition, such as dust and toxins, is essential.
- Regular Monitoring:
- Routine follow-up appointments and monitoring are necessary to assess disease progression, adjust treatment plans, and address any emerging issues.
Treatment plans should be individualized, and patients should work closely with a healthcare team, including pulmonologists and specialists, to determine the most appropriate treatment strategy. Early diagnosis and intervention can play a significant
Conclusion
Interstitial Lung Disease (ILD) and Pulmonary Fibrosis (PF) represent a group of complex and often progressive lung disorders characterized by inflammation and scarring of the lung tissue.
These conditions can significantly impact the quality of life and lung function of affected individuals. Early and accurate diagnosis, coupled with a tailored treatment plan, are essential to effectively manage these diseases and alleviate symptoms. While there is no cure for many forms of ILD and PF, ongoing research and advancements in medical therapies offer hope for improved outcomes and a better understanding of these challenging respiratory conditions.
Supportive care, multidisciplinary management, and a strong patient-provider partnership are vital components in the comprehensive approach to addressing ILD and PF, ultimately aiming to enhance the well-being of those living with these conditions.