Neuroblastoma and pheochromocytoma are two noteworthy childhood tumors that demand our attention. Neuroblastoma, a malignancy originating in nerve tissue, frequently starts in the adrenal glands, predominantly affecting young children. Pheochromocytoma, while usually non-cancerous, arises in the adrenal glands and can cause high blood pressure due to excessive hormone production. Despite their differences, understanding these tumors is vital for effective management and care.
What is Neuroblastoma?
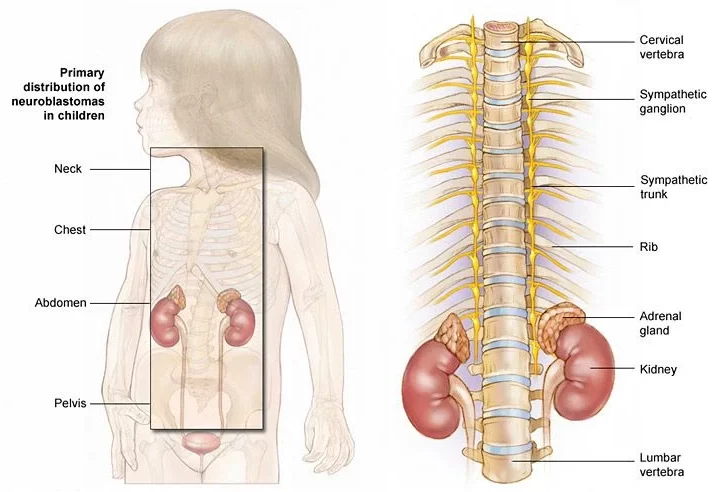
Neuroblastoma is a rare and often aggressive form of childhood cancer that originates in immature nerve cells called neuroblasts. This cancer primarily affects infants and young children, usually under the age of five. While it can develop in various parts of the body, it most commonly emerges in the adrenal glands, which sit atop the kidneys.
Neuroblastoma’s symptoms can vary widely, depending on the tumor’s size and location. Common signs include abdominal pain or swelling, a noticeable lump, weight loss, fever, irritability, and bone pain if the cancer has spread.
Genetic factors play a significant role in neuroblastoma, and specific genetic abnormalities, such as MYCN amplification, can influence the course of the disease and guide treatment decisions. Treatment approaches are comprehensive and may include surgery to remove the tumor, chemotherapy, radiation therapy, immunotherapy, targeted therapy, and stem cell transplantation.
The prognosis for neuroblastoma varies, with outcomes ranging from favorable for some patients to very poor for others, depending on factors like the child’s age, stage at diagnosis, and response to treatment. Early detection and a multidisciplinary approach to care are crucial in providing the best possible outcomes for children with neuroblastoma.
Symptoms of Neuroblastoma
Here are the symptoms of neuroblastoma:
- Abdominal swelling or mass
- Abdominal pain or discomfort
- Changes in bowel habits (constipation or diarrhea)
- Loss of appetite and weight loss
- Fatigue or lethargy
- Low-grade fever
- Bone pain or limping (if the cancer has spread to bones)
- Proptosis (bulging of one or both eyes) in rare cases
- Uncontrolled eye movements (opsoclonus-myoclonus syndrome), particularly in some subtypes of neuroblastoma
Keep in mind that the symptoms can vary from one child to another, and some children with neuroblastoma may not exhibit any symptoms until the disease has advanced. If you notice any concerning signs in a child, especially one under the age of five, it’s important to seek prompt medical evaluation for an accurate diagnosis and appropriate treatment.
Causes of Neuroblastoma
The exact causes of neuroblastoma are often unknown, but several factors are associated with its development:
- Genetic Mutations: Some cases are linked to specific genetic mutations, such as MYCN amplification, which can drive the formation of neuroblastoma.
- Hereditary Factors: In rare instances, neuroblastoma may be hereditary and run in families due to genetic predisposition.
- Spontaneous Mutations: Neuroblastoma can occur spontaneously without a known genetic cause or family history.
- Embryonic Nerve Cell Origin: Neuroblastoma originates from immature nerve cells (neuroblasts) during embryonic development.
- Age: It predominantly affects young children, with the highest incidence in infants and toddlers.
- Environmental Factors: Some research suggests potential environmental influences, but they are not well-established as direct causes.
Understanding the precise cause of neuroblastoma remains an active area of research.
What is Pheochromocytoma?
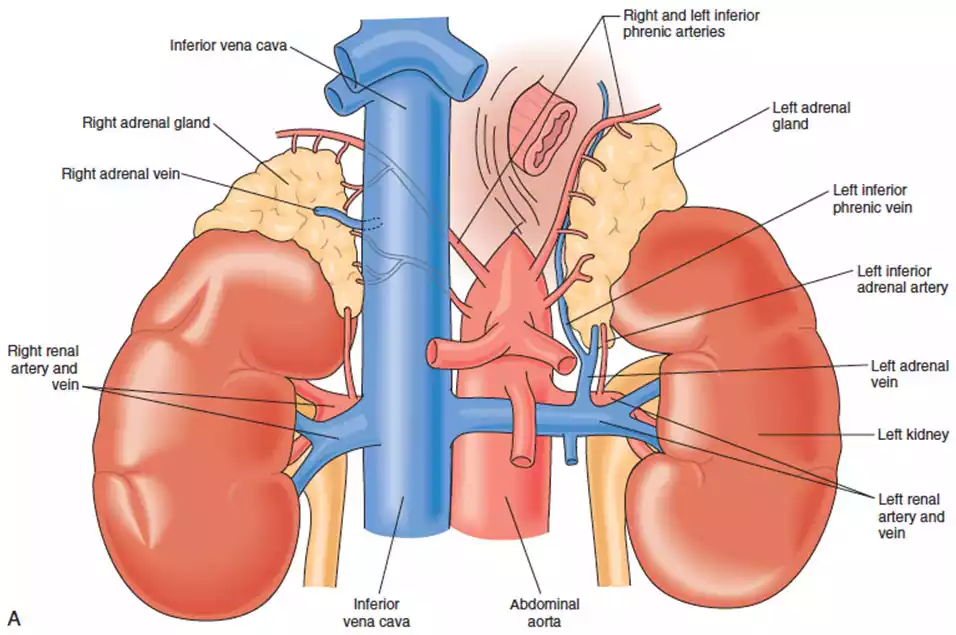
Pheochromocytoma is an extremely rare but potentially life-threatening tumor found on top of kidneys that develops from adrenal gland tissue. These tumors arise from specialized cells called chromaffin cells, which produce hormones like adrenaline and noradrenaline, also known as epinephrine and norepinephrine. Pheochromocytomas can lead to an overproduction of these hormones, causing a range of symptoms and complications.
The most common symptoms of pheochromocytoma result from excessive release of adrenaline and noradrenaline, and they can include severe high blood pressure (hypertension), headaches, rapid heart rate (tachycardia), sweating, and intense anxiety or panic attacks. In some cases, pheochromocytomas may be asymptomatic, making them challenging to diagnose without routine screening.
Treatment typically involves surgical removal of the tumor, a procedure known as adrenalectomy. Prior to surgery, medications to control blood pressure and heart rate are often administered to stabilize the patient. Pheochromocytoma can occur sporadically or as part of a genetic syndrome, such as Multiple Endocrine Neoplasia type 2 (MEN2) or Von Hippel-Lindau disease (VHL).
Regular follow-up and genetic testing may be recommended for individuals with hereditary forms to monitor for recurrence or the development of other tumors. Early diagnosis and prompt treatment are essential to prevent potentially life-threatening complications associated with pheochromocytoma.
Symptoms of Pheochromocytoma
Here are the symptoms of pheochromocytoma:
- High blood pressure (hypertension), often severe and episodic
- Headaches, which can be severe and sudden
- Rapid heartbeat (tachycardia)
- Sweating excessively, particularly during episodes of symptoms
- Paleness of the skin
- Tremors or shaking
- Anxiety or panic attacks
- Abdominal pain or discomfort
- Weight loss
- Feeling of impending doom
Pheochromocytoma symptoms are primarily due to the excess secretion of adrenaline and noradrenaline (epinephrine and norepinephrine) by the tumor. These symptoms can be intermittent and may occur in “attacks” or episodes, making diagnosis challenging. If you or someone you know is experiencing these symptoms, especially severe high blood pressure or sudden, severe headaches, it’s essential to seek immediate medical attention, as pheochromocytoma can be life-threatening if not treated promptly.
Causes of Pheochromocytoma
Pheochromocytoma primarily develops due to the following factors:
- Genetic Mutations: Specific genetic mutations, such as those affecting the RET, VHL, or NF1 genes, are associated with an increased risk of pheochromocytoma. Hereditary syndromes like Multiple Endocrine Neoplasia type 2 (MEN2), Von Hippel-Lindau (VHL) syndrome, and Neurofibromatosis type 1 (NF1) often involve these mutations.
- Sporadic Cases: Pheochromocytomas can occur spontaneously in individuals without a known genetic mutation or family history of the condition.
- Chromaffin Cell Abnormalities: Hyperplasia (excessive growth) of chromaffin cells, responsible for producing adrenaline and noradrenaline, can lead to the formation of pheochromocytomas.
- Stress and Stimulation: Stressful events or situations can stimulate the adrenal glands, causing the release of excess adrenaline and noradrenaline, potentially worsening symptoms in individuals with pre-existing pheochromocytoma.
How Are These Tumors Detected?
Detecting Neuroblastoma:
- Symptoms: Suspected based on symptoms like abdominal swelling, pain, and weight loss.
- Physical Exam: May reveal an abdominal mass.
- Imaging: Ultrasound, CT, MRI, or MIBG scans for visualizing the tumor.
- Biopsy: Tissue sampling to confirm the diagnosis.
- Blood/Urine Tests: Measure catecholamines and other markers.
- Bone Marrow Exam: To check for metastasis.
Detecting Pheochromocytoma:
- Symptoms: Suspected due to severe hypertension, headaches, and sweating.
- Blood Pressure Monitoring: Regular checks to detect hypertensive episodes.
- Hormonal Tests: Blood and urine tests for catecholamine levels.
- Imaging: CT, MRI, or MIBG scans to locate the tumor.
- Genetic Testing: Families looking back into their past should become acquainted with each other through genealogy research.
- Clonidine Suppression Test: May be used to differentiate causes of high blood pressure.
- Adrenal Venous Sampling: In some cases to determine hormone secretion and tumor location.
Early detection is crucial for effective treatment in both cases.
Imaging Techniques for Diagnosis
Common imaging techniques for diagnosis include:
- X-rays: Use ionizing radiation to create images of bones and some tissues.
- CT (Computed Tomography) Scan: Combining X-rays and computer technology, this method produces accurate cross-sectional pictures of the human body.
- MRI (Magnetic Resonance Imaging): Uses strong magnetic fields and radio waves to generate high-resolution images of soft tissues and organs.
- Ultrasound: Employs sound waves to create real-time images, often used for monitoring pregnancies and examining internal organs.
- PET (Positron Emission Tomography) Scan: Utilizes radioactive tracers to detect metabolic activity in tissues, aiding in cancer diagnosis and staging.
- MIBG (Metaiodobenzylguanidine) Scan: A specialized nuclear medicine scan used to detect neuroendocrine tumors like neuroblastoma and pheochromocytoma.
- Bone Scan: Involves injection of a radioactive substance to detect abnormalities in bone, useful for assessing bone metastasis.
- Angiography: Uses contrast material and X-rays to visualize blood vessels, helping diagnose vascular conditions.
These imaging techniques play critical roles in diagnosing a wide range of medical conditions, from fractures and tumors to vascular disorders and neurological diseases.
Treatment Approaches
Treatment Approaches for Neuroblastoma
Neuroblastoma treatment typically includes using multiple therapies concurrently:
- Surgery: To remove the tumor.
- Chemotherapy: Drugs to shrink tumors and kill cancer cells.
- Radiation Therapy: High-energy X-rays to target cancer cells.
- Stem Cell Transplantation: High-dose chemotherapy followed by stem cell infusion.
- Immunotherapy: Employing our immune systems in order to fight cancer.
- Targeted Therapy: Drugs targeting specific cancer-related molecules.
- Isotretinoin (RA Therapy): Prevents relapse in high-risk cases.
- Clinical Trials: Investigational treatments for eligible patients.
Treatment plans are tailored to the individual’s age, tumor stage, genetic markers, and response to initial therapies. A multidisciplinary team of specialists provides comprehensive care.
Treatment Approaches for Pheochromocytoma
Treatment for pheochromocytoma includes:
- Alpha and Beta-Blockers: Medications to control high blood pressure and symptoms.
- Surgical Removal: The primary treatment, removing the tumor via surgery.
- Preoperative Preparation: Medications before surgery to stabilize blood pressure.
- Genetic Testing: Identifying hereditary forms of the condition.
- Long-Term Follow-up: Regular monitoring post-surgery for recurrence.
- Metastatic Disease: Additional treatments like chemotherapy or radiation for advanced cases.
- Lifestyle Management: Avoiding triggers that stimulate adrenaline release.
A multidisciplinary approach with specialists is essential for effective management.
Comparison table of Neuroblastoma and Pheochromocytoma
Here’s a comparison table highlighting the key differences between Neuroblastoma and Pheochromocytoma:
| Characteristic | Neuroblastoma | Pheochromocytoma |
|---|---|---|
| Tumor Origin | Develops from nerve cells | Arises from adrenal gland cells |
| Age of Onset | Primarily affects children | Typically diagnosed in adults |
| Common Symptoms | – Abdominal swelling or mass
– Pain – Weight loss – Fever – Bone pain (if metastasized) |
– Severe hypertension
– Headaches – Sweating excessively |
| Genetic Factors | MYCN amplification is significant | Some cases linked to hereditary syndromes |
| Location | Often in abdomen, can occur elsewhere | Adrenal glands (usually) or extra-adrenal sites |
| Hormones Produced | May produce adrenaline and noradrenaline | Produces excess adrenaline and noradrenaline |
| Diagnostic Tests | – Imaging (CT, MRI, MIBG scans)
– Biopsy – Blood and urine tests (catecholamines) – Bone marrow examination |
– Blood and urine tests (catecholamines)
– Imaging (CT, MRI) – Genetic testing (for hereditary forms) |
| Treatment Approaches | – Surgery
– Chemotherapy – Radiation therapy – Immunotherapy – Targeted therapy – Stem cell transplantation |
– Alpha and beta-blockers
– Surgical removal (adrenalectomy) – Genetic testing – Long-term follow-up |
| Age Distribution (Common) | Typically in children under 5 years | Usually diagnosed in adults (30-50 years) |
| Prognosis | Varies widely, from favorable to poor outcomes | Generally good with appropriate treatment, poor if left untreated |
| Hereditary Forms | Some cases have genetic predisposition | Some associated with hereditary syndromes |
Individual cases may vary, and this table provides a general overview of the differences between these two conditions. Accurate diagnosis and treatment planning require consultation with healthcare professionals.
Similarities of Neuroblastoma and Pheochromocytoma
While neuroblastoma and pheochromocytoma are distinct tumors, they share some similarities:
- Neuroendocrine Origin: Both tumors arise from neuroendocrine cells.
- Catecholamine Production: They can both produce hormones like adrenaline.
- Abdominal Location: Both can develop in the abdomen.
- Hereditary Forms: Certain genetic syndromes can predispose individuals to both.
- Imaging Techniques: Similar imaging methods are used for diagnosis.
These similarities notwithstanding, they have distinct clinical features and require different management approaches.
Summary
Neuroblastoma and Pheochromocytoma are distinct neuroendocrine tumors. Neuroblastoma primarily affects children and arises from immature nerve cells, while Pheochromocytoma typically occurs in adults and originates from adrenal gland cells. Although both can produce hormones and share some imaging methods, they differ in age of onset, symptoms, and treatment approaches. Early and accurate diagnosis is crucial for the appropriate management of these rare conditions.